Phenylketonurie (PKU)
Phenylketonurie, kortweg PKU, komt voor bij 1 op de 18.000 kinderen die in Nederland worden geboren. Per jaar zijn dit er ongeveer 11. Het komt evenveel voor bij jongens als meisjes. Het is niet aan iemand te zien of degene Phenylketonurie heeft. Bij PKU is het lichaam niet of onvoldoende in staat om de phenylalanine af te breken. Dit is een stof die in het eiwit van de voeding zit. Het wordt op de verkeerde manier verwerkt waardoor phenylketonen ontstaan. De phenylketonen komen in het bloed en in de urine. Na lange tijd is dit schadelijk, vooral voor de hersenen. Phenylketonurie betekent letterlijk; phenylketonen in de urine. Mensen met PKU hebben een afgestemd eiwitarm dieet nodig.
Phenylketonurie is een ongeneeslijke erfelijke stofwisselingsziekte. De stofwisseling wordt ook wel het metabolisme genoemd. Hiermee wordt bedoeld, het verwerken en omzetten van verschillende stoffen in het lichaam. Dit is nodig voor het vrijmaken van energie. Maar ook voor de opbouw van spieren, botten en organen. Dit vindt plaats in alle cellen van het lichaam. Er bestaan ongeveer 600 verschillende soorten stofwisselingsziekten. Deze zijn altijd erfelijk en dus aanwezig vanaf de geboorte. Deze ziekten zijn zeldzaam maar hebben vaak grote gevolgen. Bij stofwisselingsziekten werkt een bepaald enzym te hard of ontbreekt het. Dit resulteert in een overschot of gebrek. Een enzym is een stof die een bepaalde lichamelijke reactie mogelijk maakt of versnelt. Enzymen zijn nodig om bepaalde voedingsstoffen om te zetten in andere stoffen die het lichaam nodig heeft. Er zijn duizenden enzymen in het lichaam aan het werk. Bij PKU mist er één enzym.
Phenylketonurie wordt veroorzaakt doordat de lever één bestanddeel van eiwitten, het aminozuur Phenylalanine (afgekort Phe, spreek uit als fee) niet of niet voldoende verwerkt. Aminozuren zijn bestanddelen van eiwitten. Verschillende soorten samen vormen een eiwit. Eiwit uit de voeding wordt in het lichaam afgebroken tot aminozuren om vervolgens gebruikt te kunnen worden. Dat Phe niet goed wordt verwerkt komt door de afwezigheid of onvoldoende aanwezigheid van een enzym. In dit geval is dat phenylalanine hydroxylase, uit de lever. Bij PKU kan phenylalanine niet omgezet worden in tyrosine. Tyrosine is een ander belangrijk aminozuur. Het aminozuur Phe hoopt zich dan op in het bloed. Bij langdurig teveel Phe in het bloed belemmert het de groei en ontwikkeling van de hersenen. En in mindere mate de ontwikkeling van het zenuwstelsel. Onbehandeld leidt PKU dan ook tot geestelijke achterstand, oftewel zwakbegaafdheid. De hoogte van het Phe gehalte in het bloed wordt maandelijks via een bloedtest gecheckt.
PKU kan, als het tijdig ontdekt wordt, goed behandeld worden. Daarom worden alle baby’s in Nederland sinds 1974 binnen acht dagen na de geboorte met de hielprik gecontroleerd op PKU/HPA. HPA of Hyperphenylalanine is een lichtere vorm van PKU. Als bij de baby PKU wordt geconstateerd, wordt meteen met de behandeling gestart.
Phenylketonurie symptomen
Bij de geboorte zijn er geen symptomen die aanduiden dat een kind PKU heeft. Door de hielprik screening wordt dit ontdekt. Er kan meestal vroegtijdig worden gestart met de behandeling. Het merendeel van de symptomen wordt daarom tegenwoordig zeer zelden tot nooit gezien. De genoemde symptomen treden vooral op bij onbehandelde patiënten. Ook de mate waarin de patiënt Phe kan verdragen, is medebepalend. Aangenomen wordt dat de meeste symptomen zich geleidelijk ontwikkelen. Dit naarmate hogere Phe-concentraties gedurende langere tijd de hersenontwikkeling nadelig beïnvloeden. Enkele symptomen kunnen zich zelfs ontwikkelen voor de geboorte, zoals een laag geboortegewicht en hartafwijkingen.
PKU-patiënten kunnen met name gaan braken, wanneer eiwitrijke voeding niet wordt verdragen. Een ander woord voor eiwit is proteïne. Geestelijke achterstand is het belangrijkste kenmerk bij onbehandelde kinderen. Met name tijdens de kinderleeftijd kunnen hoge Phe-concentraties tot onomkeerbare beschadiging van de hersenen leiden. En daarmee tot lagere IQ-niveaus. Het IQ tussen patiënten varieert, maar bijna alle onbehandelde kinderen zouden waarschijnlijk een IQ lager dan 60 hebben. Echter, bij tijdige start van de behandeling is het verschil tussen de (mentale) ontwikkeling van kinderen met PKU en die van kinderen zonder PKU klein. Ten opzichte van de algemene populatie ligt het IQ-niveau iets lager (-5 IQ-punten). En er wordt vaker speciaal onderwijs gevolgd. Maar dit is niet specifiek aan het IQ is gelinkt.
Epilepsie komt bij ongeveer een derde van de onbehandelde patiënten voor. Als epilepsie bij behandelde patiënten voorkomt is het niet het gevolg van PKU. Gedragsproblemen bij onbehandelde patiënten komen geregeld voor. Vooral in de vorm van hyperactiviteit, agressiviteit en onrust. Dit als gevolg van de verhoogde Phe-concentraties. Behandelde patiënten kunnen ook hoge Phe-concentraties hebben bij een onjuist dieet. Dit uit zich vaak in geïrriteerdheid en concentratieproblemen of depressieve klachten. PKU-patiënten hebben vaak het gevoel anders te zijn door hun ziekte en behandeling. Onbehandelde Phenylketonurie bij de moeder tijdens de zwangerschap kan leiden tot groeiachterstand bij de baby. Dit uit zich in de eerste 3 jaar van de baby.
Oorzaken phenylketonurie
Patiënten met PKU worden geboren met een gebrek aan een enzym in de lever met de naam phenylalanine hydroxylase (PAH). PAH is nodig voor het verwerken van het aminozuur phenylalanine (Phe), dat voorkomt in voedingsmiddelen die eiwit bevatten.
Overerven phenylketonurie
Phenylketonurie is erfelijk. Dat betekent dat het generatie op generatie kan worden doorgegeven. Het gebrek aan het enzym is te wijten aan een defect gen. Genen bevatten erfelijk materiaal. Erfelijke eigenschappen worden hiermee doorgegeven aan het nageslacht. De genen van de ouders vormen samen de genen van het kindje. Dit gebeurt al als de eicel bevrucht is en er een kindje in de buik van de moeder groeit. Een kind kan alleen PKU erven als beide ouders minimaal drager zijn van het defecte gen. De drie mogelijkheden zijn; drager zonder PKU (wit/grijs), drager met PKU (grijs) of geen drager (wit). De combinaties daarvan zijn als volgt. Bovenaan staan de ouders, daaronder de kansverdeling bij vier kinderen.
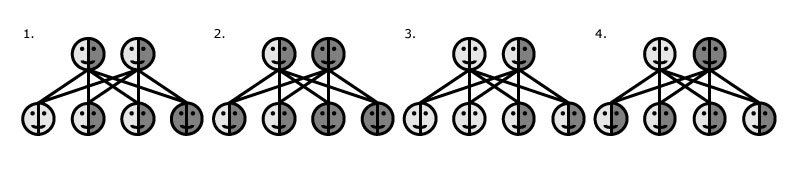
- Beide ouders zijn drager zonder phenylketonurie te hebben: Dan bestaat er een kans op PKU van 25% bij ieder kind. 50% kans om alleen drager te zijn en 25% kans om geen drager te zijn.
- Eén ouder is drager en één ouder heeft PKU: De kansen zijn nu 50/50 op het zijn van een drager en het daadwerkelijk hebben van PKU.
- Eén ouder heeft niks en één ouder is drager: Dan zijn de kansen nu 50/50 op het vrij zijn en het drager zijn.
- Eén ouder heeft niks en één ouder heeft PKU: Dan is de kans 100% dat elk kind drager wordt.
- Als beide ouders PKU hebben: Dan heeft ook elk kind PKU.
Voeding en phenylketonurie
Bij PKU volgt men een levenslang Phe beperkt dieet. Het dieet is erop gericht om de ophoping van het aminozuur Phe in het lichaam tegen te gaan. Het aminozuur is echter een onderdeel van bijna alle voedingsmiddelen doordat het in al het natuurlijk eiwit zit. Daarom zal de patiënt met PKU zijn leven lang een dieet moeten volgen dat streng beperkt (soms zeer streng beperkt) is in natuurlijk eiwit. De mensen met PKU krijgen door het eiwitbeperkte dieet veel te weinig gezonde voedingsstoffen binnen. Daarom moet naast de strenge beperking van natuurlijk eiwit, een speciaal phe-vrij preparaat ingenomen worden. Dit phe-vrije preparaat bevat alle aminozuren, vitamines en mineralen die onmisbaar zijn voor de groei van het lichaam en de ontwikkeling van de hersenen. Dit wordt vaak een aminozurenpreparaat genoemd.
Bijna alle voedingsmiddelen bevatten eiwit. In sommige producten zit zoveel eiwit dat deze nooit gegeten mogen worden. Groente, fruit en aardappelen mogen wel, maar in beperkte mate en tot op de gram nauwkeurig afgewogen. De ene patiënt kan meer eiwit verdragen dan de ander. Dagelijks moet de door de diëtist voorgeschreven hoeveelheid phe-vrij aminozuurpreparaat ingenomen worden. Afhankelijk van de phe-concentraties in het bloed wordt de dagelijkse hoeveelheid phe in het dieet vastgesteld in overleg met de diëtist.
Aspartaam is een kunstmatige zoetstof. Het wordt vaak gebruikt als suikervervanger omdat het weinig calorieën bevat. Ook aspartaam bevat eiwit en moet daarom gemeden worden. Dit zit in vele voedingsmiddelen, maar ook in medicijnen. Het is daarom aan te raden om de apotheker op de hoogte stellen.
Complicatie bij phenylketonurie dieet
Er kan door het phenylketonurie dieet een vitamine gebrek ontstaan. Dit zijn de vitamines die in eiwitrijke producten zitten. Het gaat hier om calcium, thiamine, riboflavine, vitamine B6, vitamine B12 en zink. Een tekort kan op lange termijn onder andere bloedarmoede veroorzaken. Maar tegenwoordig zijn de aminozurenpreparaten voorzien van een uitgebalanceerde hoeveelheid vitaminen en mineralen. Het aminozurenpreparaat is daarom een belangrijke aanvulling op de voeding bij PKU.
Phenylketonurie en zwangerschap
Dit wordt ook wel maternale PKU genoemd. Bij een vrouw met PKU die zwanger is of een kinderwens heeft, moeten de concentraties van phenylalanine lager liggen. Het liefst dus al vóór de zwangerschap. Dit is belangrijk om een gezond kindje te krijgen. Een te hoog gehalte kan namelijk schadelijk zijn voor het ongeboren kindje. Langdurig een te hoge Phe waarde in het bloed kan voor verschillende aandoeningen bij de baby zorgen. Bijvoorbeeld hartproblemen en een vertraagde hersenontwikkeling. Het is noodzakelijk dat de zwangere vrouw wekelijks een bloedtest uitvoert om de Phe waarde te controleren. Omdat het ongeboren kindje bloed van de moeder ontvangt.
Phenylketonurie met ziekte en koorts
Als een andere ziekte (naast PKU) of koorts langer duurt dan twee dagen is het belangrijk om contact op te nemen met de arts en diëtist. Ziekte en koorts zorgt voor een verhoogde energiebehoefte. Maar ziekte en koorts gaan vaak gepaard met een verminderde voedselinname. Ook gaat het lichaam eigen eiwit afbreken. Het gevolg is een te hoge Phe hoeveelheid in het bloed. En daarnaast heeft men nog minder energie wat nadelig werkt bij ziekte of koorts.
Externe informatie phenylketonurie of producten
We zijn extern onderstaande boeken, betreffende phenylketonurie, voor u tegengekomen:
- Nutrition During Phenylketonurie (Pku) Auteur Josef Miligui, Engels, 56 pagina’s.
- The PKU Paradox, e-BOOK. Auteurs Jeffrey P. Brosco & Diane B. Paul, Engels. The PKU Paradox is informed by interviews with scientists, clinicians, policymakers, and individuals who live with the disease. The questions it raises touch on ongoing controversies about newborn screening and what happens to blood samples collected at birth.
- 21st Century Phenylketonuria (PKU) Sourcebook: Clinical Data for Patients, Families, and Physicians – Folling’s Disease, PAH Deficiency, Sapropterin, Kuvan, Screening, Diet, Medicine, Pregnancy, e-BOOK. Engels.
- Reversing Phenylketonuria (Pku); Overcoming Cravings the Raw Vegan Plant-Based Detoxification & Regeneration Workbook for Healing Patients. Volume 3. Auteur Health Central, Engels, 106 pagina’s.
Disclaimer voedingsdoelen
DietCetera geeft u met bovenstaande tekst slechts algemene informatie. Wij hebben deze tekst niet gericht op individuele personen en omstandigheden. Vanzelfsprekend hebben we wel getracht deze informatie zo duidelijk en correct mogelijk te omschrijven. U blijft echter zelf verantwoordelijk voor uw eigen keuzes en interpretaties. Mocht u specifieke vragen of problemen hebben dan adviseren we u contact op te nemen met uw (huis)arts, diëtist of andere deskundigen. DietCetera is niet aansprakelijk voor eventuele schade ten gevolge van het onjuist interpreteren van deze tekst.
